
- Title
-
DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination
- Authors
- Wei, L., Nakajima, S., B�hm, S., Bernstein, K.A., Shen, Z., Tsang, M., Levine, A.S., Lan, L.
- Source
- Full text @ Proc. Natl. Acad. Sci. USA

DNA damage at active transcription sites induces reversible transcription inhibition by the DART system. (A) Scheme of the DART system for inflicting genome locus-specific damage. KR generates ROS upon visible light exposure and induces localized damage at transcription either off or on sites. (B and C) TA-KR transfected U2OS TRE cells were irradiated with light for 10 min followed by the indicated postlight incubation time. Phosphorylated POLII detected by anti-4H8 (B) or YFP-POLII (C) is shown at the indicated time points. Quantification of the average fold increase of 4H8 foci intensity of 10 cells at TA-KR damage sites is shown, and error bars indicate the SEM of three independent experiments. Dephosphorylation of POLII but not the dissociation of POLII was observed upon damage. (D) The percentage of CKL-CFP positive cells (defined as CFP foci > 10/cell) in tetR-KR or TA-cherry/KR transfected U2OS TRE cells before and after damage induction is shown. n = 50, and error bars indicate the SEM of three independent experiments. |

HR factors are specifically recruited to transcriptionally active damage sites. (A) Recruitment of KU70 and NBS1 at TA-KR and tetR-KR damage sites is equal after damage. (B) RAD51 is specifically recruited to TA-KR but not TA-mCherry or tetR-KR/mCherry. U2OS TRE cells were illuminated with 15W cool fluorescent white light for 10 min for damage production. The recruitment of RAD51 stained with its Ab is shown 30 min after damage production. (C) GFP tagged RPA1, RAD52, and RAD51C are specifically recruited to TA-KR with the same treatment as in A. (D) Quantification of percentage of cells with positive foci of the HR factor at the indicated spot 30 min after damage production. Error bars indicate the SEM of three independent experiments, n = 100. (E) Percentage of cells with γH2AX and 53BP1 foci (defined as the fold increase of foci at TA-KR sites >2 compared with intensity of background in the same cell) at the indicated time points after damage induction. (F) Quantification of average fold increase of foci intensity (n = 10) of the HR factor at sites of TA-KR at the indicated recovery time after damage production. Error bars indicate the SEM of three independent experiments in E and F. |
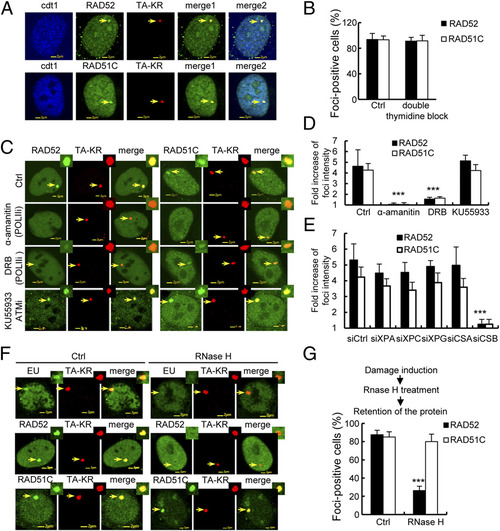
The recruitment of HR factors at TA-KR damage sites is dependent on active transcription and CSB. (A) Colocalization of RAD51C or RAD52 (green) and TA-KR (red) in cdt1 (blue) expressing U2OS-TRE cells 30 min after KR activation. (B) Quantification of the percentage of cells with positive foci at TA-KR (n = 100) in double thymidine synchronized G1 cells. Error bars indicates the SEM of three independent experiments, and the P values were determined by using Student?s two-tailed t test. (C and D) α-amanitin and DRB treatment abolish the recruitment of RAD51 and RAD51C at TA-KR. RAD52/RAD51C and TA-KR transfected U2OS TRE cells were pretreated with the polymerase II inhibitor (POLIIi) α-amanitin (100 �g/mL) for 1 h or DRB (20 �M) for 24 h or KU55933 (10 �M) for 1 h, then irradiated with light for 10 min followed by incubation for 30 min. Images and quantification of the fold increase of RAD52/RAD51C foci intensity at TA-KR damage sites are shown. (E) The fold increase of RAD52/RAD51C foci intensity of 10 cells at TA-KR damage sites is shown after siRNA treatment of the indicated protein. Data are represented as mean � SEM, and the P values were determined by using Student?s two-tailed t test. (F) RNaseH (15 units) treatment for 15 min abolishes EU incorporation and the recruitment of RAD52 at TA-KR after damage induction. Images with light irradiation for 10 min followed by 30 min incubation after RNaseH treatment are shown. (G) The percentage of cells with RAD52 or RAD51C foci of 100 cells at TA-KR damage sites is shown after RNaseH treatment. Error bars indicate the SEM of three independent experiments. |
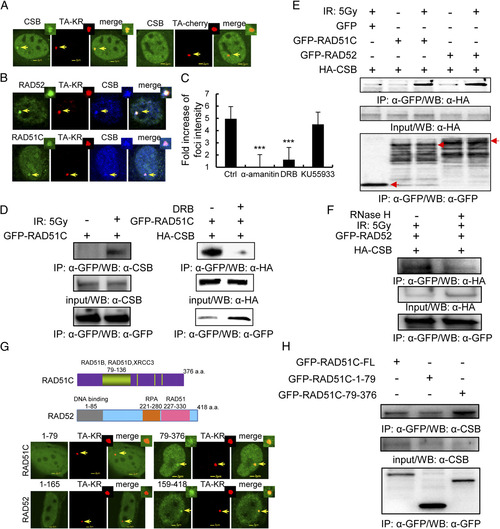
CSB localizes at TA-KR damage sites and interacts with RAD52 and RAD51C after DNA damage. (A) Recruitment of CSB at sites of TA-KR after damage. FLAG-CSB and TA-cherry/KR were cotransfected into U2OS TRE cells. Twenty-four hours later, cells were irradiated with light for 10 min followed by 30 min of incubation; the recruitment of CSB at TA-KR sites is shown with FLAG-antibody. (B) Colocalization of CSB, RAD52, or RAD51C at sites of TA-KR after damage. FLAG-CSB, GFP-RAD52/RAD51C, and TA-KR were cotransfected into U2OS TRE cells. Twenty-four hours later, cells were irradiated with light for 10 min followed by 30 min of incubation and stained with FLAG-antibody. (C) The average fold increase of FLAG-CSB at sites of TA-KR after damage (30 min postlight incubation after 10 min of light incubation) and after treatment with α-amanitin (100 �g/mL) for 1 h, DRB (20 �M) for 24 h, or KU55933 (10 �M) for 1 h. Images were quantified by ImageJ (n > 50); SEM indicates three independent experiments. (D and E) GFP-RAD52/GFP-RAD51C stably expressed in 293 cells with or without 5 Gy IR and DRB treatment was analyzed by IP with anti-GFP and 1 h postirradiation incubation. Detection of endogenous CSB or HA-tagged CSB is shown. (F) RNaseH treatment of cells abolishes the interaction between CSB and RAD52. HA-CSB expressed in GFP-RAD52 stably expressing 293 cells treated with 5 Gy IR, analyzed by IP with anti-GFP and 1 h postirradiation incubation with or without RNaseH (15 units) treatment for 15 min. Detection of HA-tagged CSB is shown. (G) The recruitment of the C-terminus RAD52 and RAD51C at TA-KR 30 min after KR activation. (H) CSB interacts with the C terminus of RAD51C. GFP-RAD51C fragments were transiently transfected into 293 cells. Forty-eight hours after transfection, cells were irradiated with 5 Gy IR. Cell lysates were collected 1 h after irradiation and immunoprecipated by GFP/HA antibody. Western blotting by anti-HA, anti-GFP, and anti-CSB for immunoprecipitates and input are shown. |

The recruitment of CSB at TA-KR damage sites is via its C terminus and depends upon its ATPase activity. (A) Scheme of CSB mutants and deletions used in the study. (B) CSB aa 337?1493 is recruited to sites of TA-KR after damage. GFP-RAD52 or RAD51C, CSB or the indicated CSB mutant, and TA-KR were transfected into siCSB pretreated U2OS-TRE cells. The recruitment of GFP-RAD52 or RAD51C (green) and CSB (blue) at TA-KR is shown 30 min after KR activation. (C) CSB aa 337?1493 complements the recruitment of RAD51C and RAD52 after damage. The fold increase of foci intensity of GFP-RAD52 or RAD51C at TA-KR damage sites is quantified (n = 10). (D) The recruitment of GFP-RAD52 or RAD51C (green) and CSB K538R or Ub? (blue) at TA-KR is shown 30 min after KR activation. (E) CSB UbΔ but not K538R complements the recruitment of RAD52 after damage. The fold average increase of foci intensity of GFP-RAD52 at TA-KR damage sites is quantified (n = 10). (F and G) GFP-RAD52/GFP-RAD51C stably expressed in 293 cells with or without 5 Gy IR was immunoprecipitated by anti-GFP with 1 h postirradiation incubation. Detection of CSB deletions (F) or mutants (G) by anti-FLAG, HA, or myc is shown. |
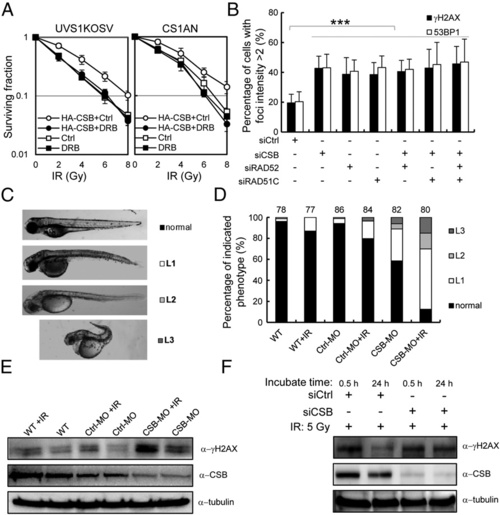
CSB is important for cell survival and in vivo after damage coupled with active transcription. (A) UVs1KOSV and HA-CSB expressing UVs1KOSV cells (Left) and CS1AN and HA-CSB expressing CS1AN cells (Right) were pretreated with or without DRB (20 �M) for 24 h, then irradiated with IR at the indicated dose. Colony forming assays were performed, and the survival fraction is shown. (B) Persistent γH2AX and 53BP1 signals at sites of TA-KR after damage without HR and/or CSB. U2OS TRE cells were treated with the indicated siRNA, then transfected with TA-KR. Cells were exposed to white light for 10 min followed by 48 h of postlight incubation, then fixed and immunostained with anti-γH2AX and anti-53BP1. The percentage of γH2AX and 53BP1 foci postive cells (defined as the fold increase of foci at TA-KR sites compared with background intensity being >2) was quantified. (C) Zebrafish embryos injected with CSB-MO/con-MO or WT were irradiated with or without 5 Gy IR at 6 h postfertilization (hpf). Images of three levels of abnormalities (L1, L2, and L3) at 72 hpf are shown. (D) Percentage of embryos in each of the categories. Error bars indicate the SEM of three independent experiments, and the P values were determined by using Student?s two-tailed t test. (E and F) Western blot of CSB and γH2AX in embryos in D (E) or in siCSB treated 293 cells with or without irradiation (F). EXPRESSION / LABELING:
PHENOTYPE:
|
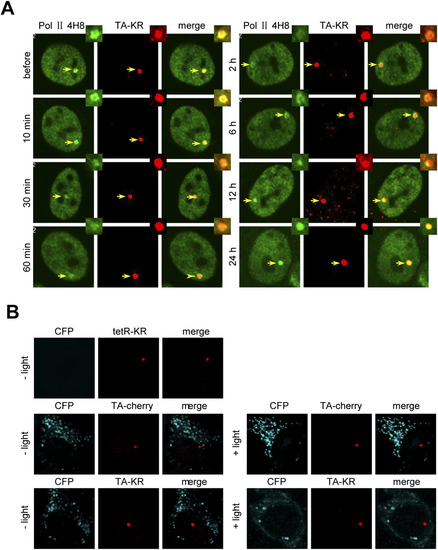
Transcription blocked by damage recovers 24 h after damage induction. (A) TA-KR transfected U2OS TRE cells were irradiated with light for 10 min followed by the indicated postlight incubation time. Phosphorylated POLII detected by anti-4H8 at the indicated time points is shown. (B) Transcription at tetR-KR or TA-cherry/KR transfected U2OS TRE cells before and after damage induction was assessed. Images of CKL-CFP expression under confocal microscopy are shown. |
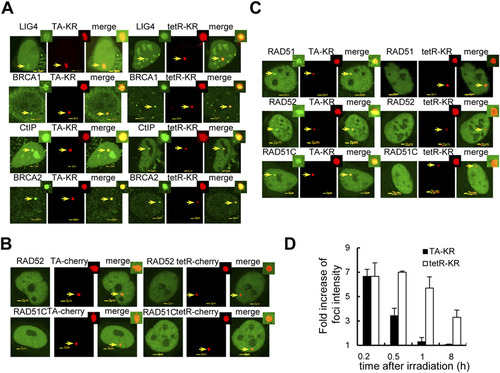
The HR factors are specifically recruited at transcriptionally active damage sites in both U2OS TRE and U2OS 263 cell lines. (A) Recruitment of LIG4, BRCA1, CtIP, and BRCA2 at TA-KR and tetR-KR damage sites in U2OS TRE cells 10 min after light irradiation is shown. (B) U2OS TRE cells were cotransfected with GFP tagged RAD52 or RAD51C and tetR-cherry or TA-cherry; 24 h after transfection, cells were illuminated with fluorescent light for 10 min. There is no recruitment of these factors at sites of tetR-cherry or TA-cherry. (C) The recruitment of the HR factors in U2OS 2-6-3 cells. U2OS 263 cells were cotransfected with GFP tagged RAD52, or RAD51C and tetR-cherry/KR or TA-cherry/KR; 24 h after transfection, cells were illuminated with fluorescent light for 10 min. RAD51 was stained with antibody. The recruitment of these factors specifically at sites of TA-KR is shown. (D) Quantification of average fold increase of foci intensity (n = 10) of the NHEJ factor at sites of tetR-KR or TA-KR at the indicated recovery time after damage. Error bars indicate the SEM of three independent experiments. |
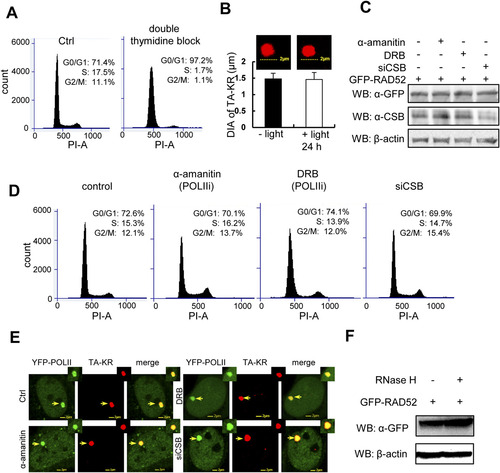
Cell cycle-independent and CSB-dependent recruitment of the HR factors at TA-KR damage sites. (A) U2OS TRE cells were synchronized at the G1 phase by double thymidine block and stained with PI. The graph shows distribution of the cell population by FACS. (B) TA-KR transfected U2OS TRE cells with/without light irradiation 24 h postincubation. Diameter of TA-KR was assessed by confocal microscopy. (C?E) Western blot of GFP-RAD52 and CSB (C), cell cycle profiles (D), and recruitment of YFP-POLII (E) in U2OS TRE cells pretreated with the RNA POLII inhibitor α-amanitin (100 �g/mL) for 1 h, DRB (20 �M) for 24 h, or siCSB. D shows FACS analysis of U2OS TRE cells with the same treatment as in C. (F) Western blot of GFP-RAD52 in cells with or without RNaseH treatment (15 units) for 15 min. |
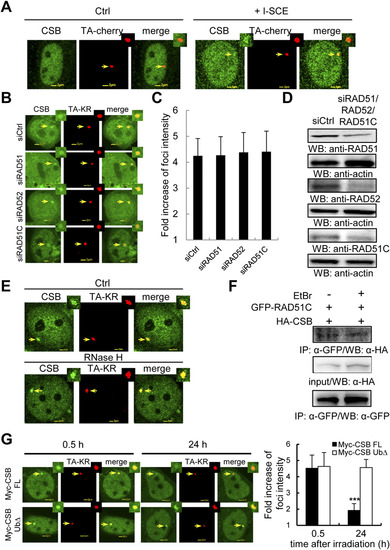
CSB recruited at transcriptionally active damage sites is independent of HR factors. (A) CSB is recruited to I-SCEI induced DSBs. TA-cherry and HA-CSB with/without I-SCEI were cotransfected into U2OS TRE cells. After 24 h, cells were fixed and stained with HA-antibody. The recruitment of CSB at TA-cherry with/without I-SCEI sites is shown. (B) siRAD51, siRAD51C, or siRAD52 transfected U2OS TRE cells were expressed with FLAG-CSB and TA-KR. The recruitment of CSB at sites of TA-KR is shown after light irradiation for 10 min followed by 30 min of incubation. (C) The fold increase of CSB intensity after knockdown of the indicated protein. (D) Knockdown effects of RAD51, RAD52, and RAD51C by siRNAs. Western blot of siCtrl, siRAD52, or siRAD51C transfected U2OS TRE cells with the indicated antibody. (E) The recruitment of CSB at sites of TA-KR in U2OS TRE cells with light irradiation for 10 min followed with 30 min incubation. Cells were pretreated for 15 min with RNaseH (15 units). (F) RAD51C interacts with endogenous CSB independently of DNA after DNA damage. Flp-in 293 cells stably expressing GFP-RAD51C were treated with 5 Gy IR with 1 h postirradiation incubation. Then cell lysates with/without 10 �g/mL EtBr were applied for IP by anti-GFP. Detection of endogenous CSB is shown. (G) Myc-CSB FL or Ub? and TA-KR cotransfected U2OS TRE cells were irradiated with light for 10 min followed by 0.5-24 h incubation. Cells were immunostained by anti-Myc. Data are represented as mean � SD, and the P values were determined by using Student?s two-tailed t test. |
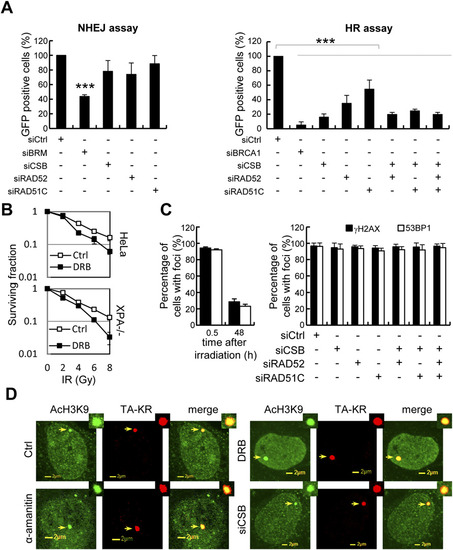
NER-independent role of CSB in repair of strand breaks at a transcribed region. (A) Effects of siRNA for the indicated protein for HR and NHEJ. (B) HeLa and XP12RO (XPA-/-) cells were pretreated with or without DRB (20 γM) for 24 h, then irradiated with IR at the indicated dose. Colony forming assays were performed, and the survival fraction is shown.(C) U2OS TRE cells with FBS starvation were treated with or without the indicated siRNA, then transfected with TA-KR. Cells were irradiated with light for 10 min followed by 30 min/48 h of incubation, then fixed and immunostained with anti-γH2AX and anti-53BP1. The percentage of γH2AX and 53BP1 foci positive cells (defined as the fold increase of foci at TA-KR sites compared with background intensity being >2) was quantified. (D) α-amanitin (100 �g/mL) for 1 h, DRB (20 �M) for 24 h, or siCSB pretreated cells were fixed, followed by immunostaining with anti-AcH3K9. |