
- Title
-
Systems Analysis of Biliary Atresia Through Integration of High-Throughput Biological Data
- Authors
- Min, J., Ningappa, M., So, J., Shin, D., Sindhi, R., Subramaniam, S.
- Source
- Full text @ Front. Physiol.
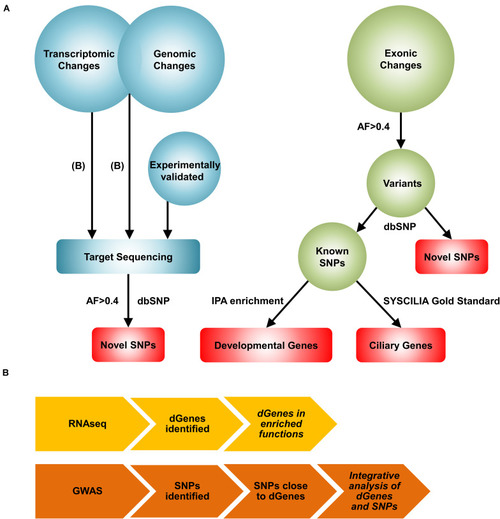
A novel systems biology approach for the reconstruction of the BA network. |
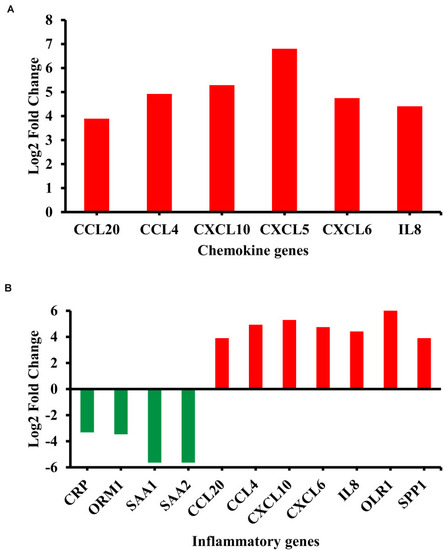
Differentially regulated genes in enriched biological categories. |
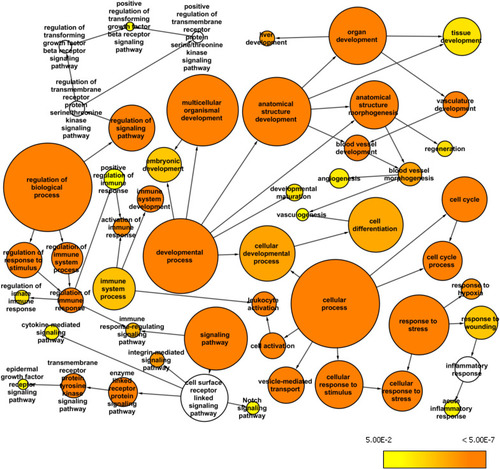
Whole exome network. This network was created in Cytoscape with the BINGO plugin to visualize over-represented Gene Ontology: Biological Processes among the genes mapped from the common variants from the whole exome data (AF>0.2 and AN>10) and their first neighbor genes within the custom human interaction network. The size of a node represents the number of genes annotated with that biological process while the color indicates different |
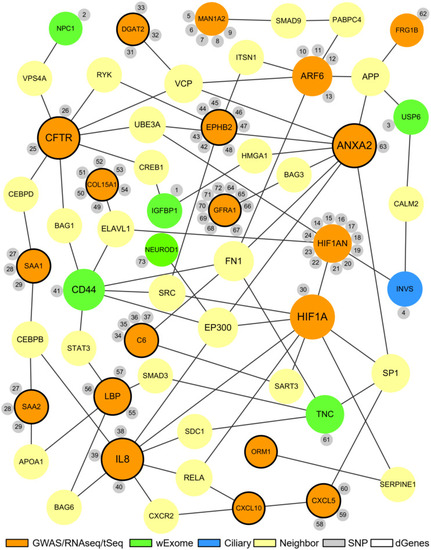
The proposed biliary atresia network. This network was created in Cytoscape by using the second neighbor interactions of the significant genes within the custom human interaction network. Interaction with other significant genes was the key criteria for selection into the final BA network. The size of a node depends on the connectivity within the network; the larger the node, the more connected it is to other genes. The orange nodes represent the significant genes derived from the GWAS, RNAseq, or target sequencing data while the light green nodes represent the significant genes from the whole exome data. One blue ciliary gene is also from the whole exome data. The yellow nodes represent the neighbor genes that link different significant genes through protein-protein interaction. The small gray nodes represent the SNPs that are associated with their attached genes. The entire list of SNPs, both novel and known, can be found in the |
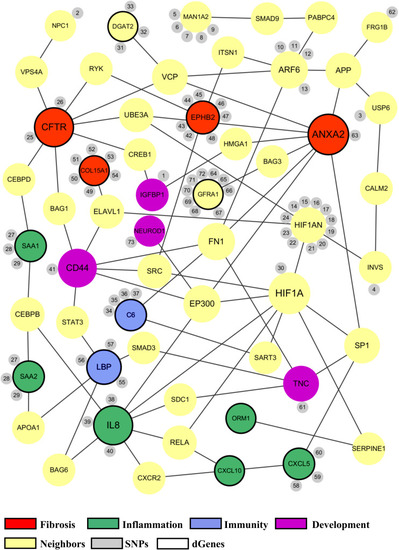
Common biological functions in the proposed biliary atresia network. All of the significant genes in the proposed BA network were annotated to identify common biological functions within the network. The red nodes represent the genes related to fibrosis, green related to inflammation, blue related to immune response, and purple related to development. The size of a node depends on the connectivity within the network; the larger the node, the more connected it is to other genes. The yellow nodes represent the neighbor genes that link different significant genes through protein-protein interaction. The small gray nodes represent the SNPs that are associated with their attached genes. The entire list of SNPs, both novel and known, can be found in the |
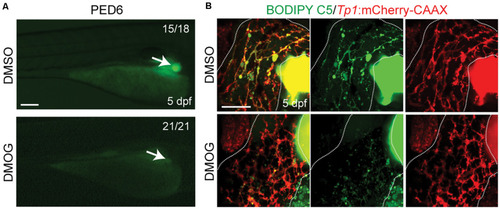
Activation of Hif1a signaling impairs biliary morphogenesis. |
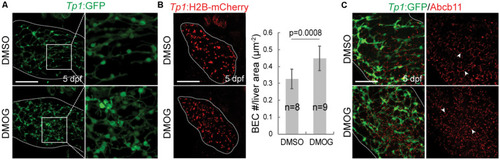
Activation of Hif1a signaling increases BEC number and impairs the proper formation of bile canaliculi. |