PRODUCTION OF ANDROGENETIC HAPLOIDS AND DIPLOIDS
(Source: G.E. Corley-Smith, C.J. Lim, and B.P. Brandhorst, submitted for publication)
Overview of procedure:
Androgenetic haploids can be produced by holding zebrafish eggs in coho salmon ovarian fluid, irradiating them with 10,000 R of x-rays, and then fertilizing them with normal sperm. To produce diploid androgenotes, the same procedure is followed as for haploid androgenotes, but is followed by inhibition of the first mitotic division. The first mitotic division can be inhibited by various methods. We use heat shock because it is easy to perform and control.
Equipment Required to Produce Haploid Androgenotes:
1. Irradiation source. If using an x-ray source, it should produce at least 150 KV. We purchased a Torrex 150D cabinet style x-ray inspection system (Faxitron X-Ray Corp., Buffalo Grove, IL., USA. phone: 708 465-9729). Based on research we have performed using salmon eggs, we believe gamma rays should also work well for zebrafish eggs.
2. In vitro fertilization supplies (see
Delayed In Vitro Fertilization Using Coho Salmon Ovarian Fluid)
Additional Equipment Required to Produce Diploid Androgenotes:
1. Hot water bath. Set to maintain fish water in a beaker at 28.5+/-0.5C. Fish water is a term we apply to the water we use for raising fish. If desired, embryo water (refer to Recipe Section of this book) can be used in this protocol, wherever fish water is indicated. Since timing of the first mitotic division is temperature dependent, accurate temperature control of this hot water bath is particularly important for producing diploid androgenotes.
2. Second hot water bath. Set to maintain fish water in a beaker at 41.4+/-0.05C. A calibrated thermometer is required for this accuracy (e.g. Fisher Scientific, Cat. No. 15041A). Inside both hot water baths we have beakers containing fish water. To promote heat transfer, the water in the beakers is constantly stirred using either a stir paddle or a magnetic stir bar. Temperatures should be measured in the beakers, since water temperatures in the beakers are usually slightly lower than in the surrounding water baths due to thermal loss from evaporation.
3. Heat Shocking Tubes. To transfer eggs between hot water baths and allow for abrupt thermal changes to be applied to the eggs, we use uncapped 50 ml polypropylene conical tubes from which the bottoms have been sliced off and a fine mesh melted on. We use 153um pore size Nitex mesh on these heat shocking tubes.
Collection of Eggs:
We collect eggs into approximately 100ul coho ovarian fluid located in the center of a 50mm diameter petri dish.
Irradiation of Eggs:
We irradiate eggs at 23 cm from the focal point of the x-ray beam of the Torrex 150D (shelf 8). Settings used are 145 KV and 5 mA. X-ray dosimetry indicates that at this distance and electrical setting, the x-ray output is 12.2 R/sec. Thus, we irradiate for 820 sec to achieve a total dose of 10,000R. Eggs are irradiated in coho ovarian fluid at room temperature. We attempt to have a monolayer of eggs with as thin a layer of ovarian fluid over eggs as possible. The Torrex 150D has a built in 1.2mm beryllium window. We use no extra filters, as they extend the time required to deliver 10,000R. If an x-ray machine with sufficient output is used, a 0.5mm aluminum or copper filter will help to selectively remove soft x-rays. Soft x-rays (low KeV) are suspected of causing more cytological damage than hard x-rays which are more selective in targeting DNA.
Fertilization of Eggs and Production of Haploid Androgenotes:
Proceed as described in Delayed In Vitro Fertilization Using Coho Salmon Ovarian Fluid.
Embryos that subsequently develop and exhibit the haploid syndrome are putative haploid androgenotes. No embryos having diploid appearance should be observed.
Heat shocking to produce diploid androgenotes:
1. Start timer as soon as 0.5 ml of 28.5+/-0.5C fish water is added to milt and eggs. This is time = 0.0 minutes.
2. Place petri dish in a 28.5C incubator or on a shallow ledge in a beaker containing 28.5C water. After 1 minute, very gently add 28.5C water to 3/4 full petri dish. 3. At 5 min, transfer eggs to a heat shocking tube (50ml tube with net bottom) and suspend tube in beaker containing 28.5C fish water that is in water bath. Tubes should be suspended, not rested on bottom of beakers and should be left uncapped.
4. At 13 min, transfer heat shocking tube containing eggs to beaker containing 41.4C fish water.
5. At 15 min, very gently transfer heat shocking tube containing eggs back to 28.5C beaker and leave there undisturbed for 1.5 h.
6. After 1.5 h, transfer eggs very gently into petri dishes 3/4 full of water and place in a 28.5C incubator.
7. At 24 h, view developing embryos under dissecting microscope. At 24 hours, if many haploid and no diploid embryos are observed in the irradiated and nonheat shocked group, any embryos in the irradiated and heat shocked group that have a diploid appearance should be diploid androgenotes.
Assessing Putative Haploid and Diploid Androgenotes:
When attempting to produce diploid androgenotes, it is advantageous to have at least three groups of eggs: 1) normal diploid control group; 2) irradiated and not heat shocked (putative haploid androgenotes); 3) irradiated and heat shocked (putative diploid androgenotes).
After collecting eggs, put a small group of eggs aside (control group) and irradiate the rest. Fertilize all eggs at same time. Part of the irradiated group can go into 28.5C incubator (potential haploid androgenotes), and the rest are heat shocked (potential diploid androgenotes). The control group is to ensure that delayed in vitro fertilization is working and to allow for the visual comparison of putative haploid and diploid androgenetic embryos with normal diploid embryos, as well as for genetic analysis.
Two phenotypes that must be recognizable by the researcher are the diploid phenotype and the haploid phenotype. Haploid embryos exhibit what is called the haploid syndrome: shortened body, small melanocytes. The haploid syndrome can be seen at 24 hours as a shortened body phenotype (Figure 1). At 48 hours, the shortened body is easily noticeable and the difference in melanocyte size starts to become noticeable (Figure 1) and is pronounced by 96 hours (not shown). The development of androgenetic diploid embryos is initially retarded in relation to diploid control embryos (Figure 1). However, by the end of the first month, the androgenetic diploids achieve approximately the same size as the diploid control fish.
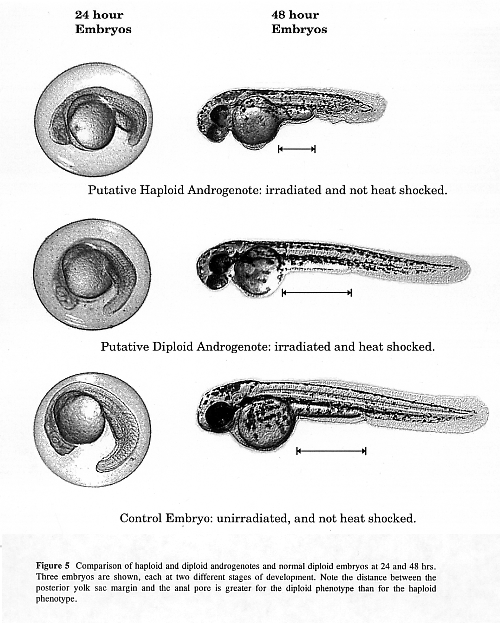
We originally determined the irradiation dosage based on the Hertwig effect (Hertwig, 1911). To ensure that the irradiation dose is adequate to destroy the maternal genome in each experiment, we always include an irradiated and non-heat shocked group. No diploid phenotypes should ever be observed in this group. If no diploid phenotypes are observed in the irradiated and non-heat shocked group, then it is likely that diploid phenotypes in the irradiated and heat shocked group are androgenetic diploids.
Confirmation of exclusive paternal inheritance requires investigating the inheritance of parentally polymorphic DNA markers to putative androgenetic progeny. The lack of homozygous maternally specific markers in the progeny is strong evidence supporting sole paternal inheritance, although it does not rule out the possibility of some maternal leakage.
References: Hertwig, O. (1911) Die Radiumkrankheit tierischer Keimzellen. Arch. Mikr. Anat. 77:1-97.
Acknowledgments: We are indebted to Charline Walker for the *AB line of fish, and for her extremely helpful advice on in vitro fertilization and on heat shocking zebrafish eggs.
The Zebrafish Book
