
- Title
-
Regenerating zebrafish fin epigenome is characterized by stable lineage-specific DNA methylation and dynamic chromatin accessibility
- Authors
- Lee, H.J., Hou, Y., Chen, Y., Dailey, Z.Z., Riddihough, A., Jang, H.S., Wang, T., Johnson, S.L.
- Source
- Full text @ Genome Biol.
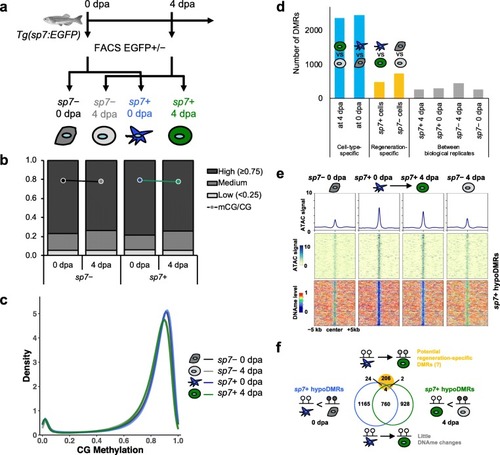
Lineage-specific DNA methylation signatures are stably maintained during fin regeneration. |
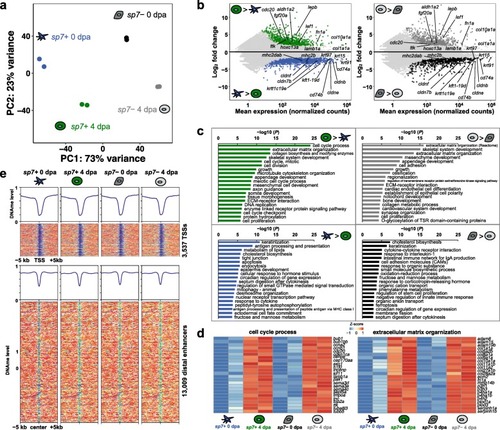
Regeneration-specific genes are activated independent of DNA methylation changes. |
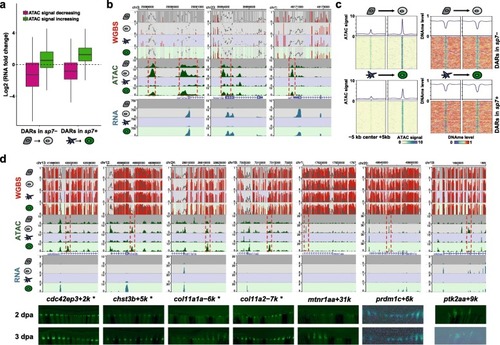
Regeneration-specific gene activation with gain of chromatin accessibility. EXPRESSION / LABELING:
|
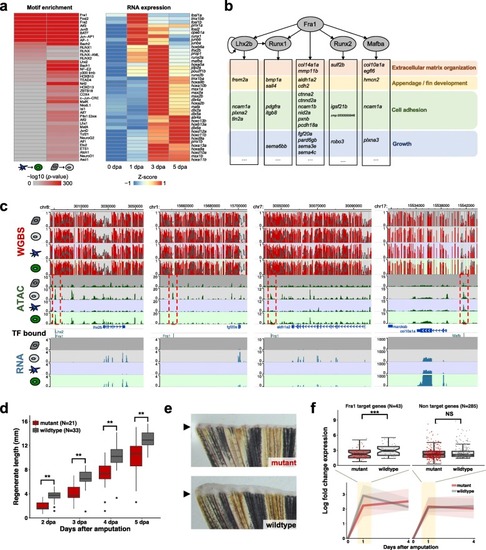
Gene regulatory networks identify upstream factors for fin regeneration. PHENOTYPE:
|